
Perfil clínico y estrategias terapéuticas en pacientes con miocardiopatía arritmogénica tratados en un instituto de referencia nacional
DOI:
https://doi.org/10.47487/apcyccv.v2i1.123Palabras clave:
Taquicardia ventricular, Muerte súbita, Displasia arritmogénica de ventriculo derechoResumen
Objetivo. Determinar las características epidemiológicas, clínicas, electrocardiográficas, imagenológicas y principales estrategias terapéuticas realizadas en los pacientes con cardiomiopatía arritmogénica tratados en un instituto cardiovascular de referencia nacional.
Materiales y métodos. Estudio observacional, descriptivo y retrospectivo que busca identificar las características clínicas, exámenes complementarios y estrategias terapéuticas en pacientes con cardiomiopatía arritmogénica tratados en el Instituto Nacional Cardiovascular – INCOR EsSalud en Lima – Perú.
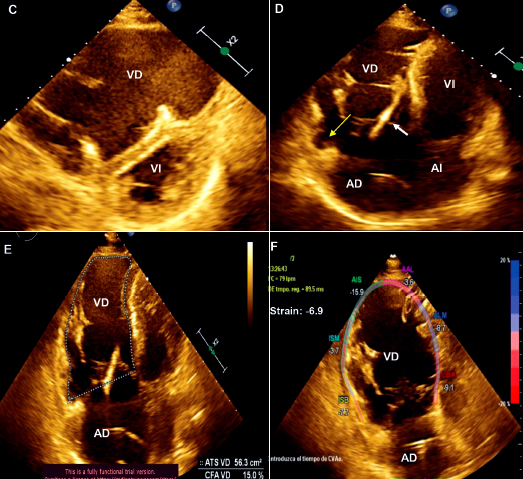
Resultados. Se encontraron trece pacientes con cardiomiopatía arritmogénica. La mediana de edad en la que se realizó el diagnóstico fue 38,2 años y el 69,3% de los afectados era de sexo masculino. Las manifestaciones clínicas más frecuentes fueron las palpitaciones taquicárdicas (92,3%), el presíncope (84,6%) y la falla cardiaca (69,2%). El 23% de los pacientes sufrió un arresto cardiaco. Todos presentaron, al menos, un episodio de taquicardia ventricular, el 92,3% con morfología de bloqueo completo de rama izquierda y eje superior. Al 76,9% se le colocó un desfibrilador automático implantable (DAI), al 15,3% se le realizó ablación y al 15,3% un trasplante cardiaco. El 84,6% de los pacientes vive hasta la actualidad.
Conclusiones. La cardiomiopatía arritmogénica afectó predominantemente a población joven y de sexo masculino. Todos los pacientes tuvieron una arritmia ventricular potencialmente fatal. La afección biventricular por ecocardiografía y por resonancia magnética cardiaca se evidenció en el 69,2 y 100% de los casos, respectivamente. Las estrategias terapéuticas empleadas fueron el tratamiento médico antiarrítmico, colocación de un DAI como prevención secundaria, la ablación y el trasplante cardíaco. Hasta la actualidad, el 84,6% de los pacientes sobrevive.
Descargas
Referencias
Gandjbakhch E, Redheuil A, Pousset F. et al. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Journal of the American college of cardiology 2018;72(7):784–804. DOI: https://doi.org/10.1016/j.jacc.2018.05.065
Cox M, Zwaag P, Van der W. et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy clinical perspective: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/ cardiomyopathy genotype-phenotype follow-up study. Circulation 2011; 123:2690–700. DOI: 10.1161/CIRCULATIONAHA.110.988287
Calkins H. Arrhythmogenic right ventricular dysplasia/cardiomyopathy-three decades of progress. Circulation Journal. 2015;79(12): 901–13. DOI-. 10.1253/circj.CJ-15-0288
Li K, Bazoukis G, Liu T et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) in clinical practice. Journal of Arrhythmia. 2018; 34:11–22. DOI: 10.1002/joa3.12021
Weijia Wang C, Calkins H. Diagnostic and therapeutic strategies for arrhythmogenic right ventricular dysplasia/cardiomyopathy patient. Europace (2019) 21, 9–21. DOI: 10.1093/europace/euy063
Corrado D, Link M, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med 2017; 376:61-72. DOI: 10.1056/NEJMra1509267
Gupta R, Tichnell C, Murray B, Rizzo S, Riele AT, Tandri H, et al. Comparison of features of fatal versus nonfatal cardiac arrest in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol 2017;120:111–7. DOI: 10.1016/j.amjcard.2017.03.251
Te Riele AS, James CA, Philips B. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol 2013;24:1311–1320. DOI: 10.1111/jce.12222
Riele T, Bhonsale A, James C, Rastegar N, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1761–9. DOI: 10.1016/j.jacc.2012.11.087
Abrams D, Kirkby C, Page S, et al. Evolution of electrocardiographic and structural features over 3 decades in arrhythmogenic cardiomyopathy. Circulation 2015; 131:2233–5. DOI: 10.1161/CIRCULATIONAHA.115.014371
Jain R, Dalal D, Daly A, Tichnell C, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia. Circulation 2009; 120:477. DOI: 10.1161/CIRCULATIONAHA.108.838821
Andreu D, Fernández –Armenta J, Acosta J, et al. A QRS axis-based algorithm to identify the origin of scar –related ventricular taqhycardia in the 17- segment American Heart Association model. Heart Rhythm.2018;15(10): 1491-1497. DOI: 10.1016/j.hrthm.2018.06.013
Corrado D, Basso C, Judge D. Arrhythmogenic cardiomyopathy. Circ Res 2017; 121:784–802. DOI: 10.1161/CIRCRESAHA.117.309345
Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation 2006;113:1634–1637. DOI: 10.1161/CIRCULATIONAHA.105.616490
Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007;115:1710–1720. DOI: 10.1161/CIRCULATIONAHA.106.660241
Sen-Chowdhry S, Syrris P, Prasad S, et al. Left-dominant arrhythmogenic cardiomyopathy: an underrecognized clinical entity. J Am Coll Cardiol 2008;52:2175–2187. DOI: 10.1016/j.jacc.2008.09.019
Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovascular Pathology 14 (2005) 37 – 41. DOI: 10.1016/j.carpath.2004.12.001
Basso C, Ronco F, Marcus F, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogewe3nic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J 2008;29:2760–2771. DOI: 10.1093/eurheartj/ehn415
Te Riele A, Tandri H, Bluemke D. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson 2014;16:50. DOI: https://doi.org/10.1186/s12968-014-0050-8
Perazzolo M, Rizzo S, Bauce B, et al. Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz 2015; 40:600–606. DOI: 10.1007/s00059-015-4228-0
Marcus FI, McKenna WJ, Sherrill D, Basso C, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 2010; 121:1533–1541. DOI: 10.1161/CIRCULATIONAHA.108.840827
Aquaro GD, Barison A, Todiere G, et al. Usefulness of combined functional assessment by cardiac magnetic resonance and tissue characterization versus task force criteria for diagnosis of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2016;118:1730–1736. DOI: 10.1016/j.amjcard.2016.08.056
Taylor AJ, Cerqueira M, Hodgson JM, et al. ACCF/SCCT/ACR/AHA/ASE/ASNC/NASCI/SCAI/SCMR 2010 appropriate use criteria for cardiac computed tomography. J Am Coll Cardiol 2010;56:1864–94. DOI: 10.1016/j.jacc.2010.07.005
Te Riele AS, Tandri H, Sanborn DM, Bluemke DA. Noninvasive multimodality imaging in ARVD/C. J Am Coll Cardiol Img 2015;8:597–611. DOI: 10.1016/j.jcmg.2015.02.007
Kimura F, Sakai F, Sakomura Y, et al. Helical CT features of arrhythmogenic right ventricular cardiomyopathy. RadioGraphics 2002;22:1111–24. DOI: 10.1148/radiographics.22.5.g02se031111
Calkins H, Corrado D, Marcus F. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2017;136: 2068–2082. DOI: 10.1161/CIRCULATIONAHA.117.030792
Li K, Bazoukis G, Liu T et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) in clinical practice. Journal of Arrhythmia. 2018; 34:11–22. DOI: 10.1002/joa3.12021
Donato G, De Luca A, Cappelletto C, et al. Prognostic Value of Magnetic Resonance Phenotype in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. J Am Coll Cardiol 2020; 75:2753–65. DOI: 10.1016/j.jacc.2020.04.023
Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 2010;122: 1144–52. DOI: 10.1161/CIRCULATIONAHA.109.913871
Bhonsale A, Groeneweg J, James C, et al. Impact of genotype on clinical course in arrhythmogenic riight ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J 2015; 36:847–55. DOI: 10.1093/eurheartj/ehu509
Rigato I, Bauce B, Rampazzo A, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet 2013;6: 533–42. DOI: https://doi.org/10.1161/CIRCGENETICS.113.000288
Elliott P, Anastasakis A, Asimaki A, et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. European journal of heart failure 2019; 21(8): 955–64. DOI: https://doi.org/10.1002/ejhf.1534
Descargas
Publicado
Número
Sección













