Cardiopatías
congénitas asociadas a los síndromes cromosómicos más prevalentes: revisión de
la literatura
Congenital heart disease associated with the most
prevalent chromosomal syndromes: a literature review
José
Eduardo Castillo Lam1,a
https://orcid.org/0000-0002-2445-7780
Oscar
Eduardo Elías Adauto 1,a
https://orcid.org/0000-0002-5070-9128
Gian
Paolo Huamán Benancio2,b
https://orcid.org/0000-0002-5426-1842
DOI:
https://doi.org/10.47487/apcyccv.v2i3.155
Resumen
Los síndromes cromosómicos más
frecuentes (Down, Patau, Edwards, Turner y Williams)
afectan de diversas formas a la población pediátrica, y las cardiopatías congénitas
asociadas explican la compleja calidad de vida que padecen; sin embargo, no se
cuenta con muchos estudios que hagan una revisión acerca de las principales
anomalías cardiacas en estos síndromes, y los que existen son publicaciones de
décadas pasadas. Se hizo una revisión en bases de datos como MEDLINE, LILACS,
SCIELO y Google Scholar, para seleccionar la mejor
evidencia posible; cada síndrome cromosómico se investigó en relación con
cardiopatías congénitas, por separado, y se organizaron cinco grupos de
búsqueda. El artículo muestra las características de cada una de las
cardiopatías asociadas que fueron descritas en los estudios revisados,
información relevante de cada estudio (autor, fecha de publicación, país y
población estudiada), además de una breve descripción que consigna la
frecuencia de la cardiopatía y su mortalidad. Se contrastaron los resultados
descritos en esta revisión, con literatura previa ya existente con la finalidad
de verificar la existencia de correspondencia entre las frecuencias reportadas.
Las cardiopatías congénitas más frecuentes fueron: defecto septal atrioventricular (DSAV), comunicación interventricular
(CIV), comunicación interauricular (CIA) y persistencia de conducto arterioso
(PCA) en el síndrome Down; PCA, CIA y CIV en el síndrome Patau;
DSAV, PCA y defectos multivalvulares en el síndrome
Edwards; aorta bicúspide, coartación de aorta y estenosis aórtica en el
síndrome de Turner, y estenosis aórtica supravalvar y
estenosis pulmonar en el síndrome Williams.
Palabras
clave:
Cardiopatías congénitas; Síndrome de Down; Síndrome de la trisomía 13; Síndrome
de la trisomía 18; Síndrome de Turner;
Síndrome de Williams (fuente: DeCS BIREME).
Abstract
Most frequent chromosomal syndromes like Down, Patau, Edwards, Turner, and Williams affect
the pediatric population in various ways, and congenital heart disease explains
the altered quality of life they suffer. There is a lack
of studies reviewing the cardiac
anomalies in these syndromes, and the ones that exist
are publications from past decades. We
reviewed databases such as MEDLINE, LILACS, SCIELO, and Google Scholar, selecting the best possible
evidence, and each chromosomal syndrome was investigated in relation to congenital
heart disease, constituting five search groups. The article shows the characteristics of each heart
disease described in the studies reviewed,
the author, date of publication, country, and population studied, as well as a brief description of the frequency of
the disease and its mortality. The results described
in this review were contrasted with previous existing
literature to verify if there
was correspondence between the reported
frequencies. The most frequent congenital
heart diseases were atrioventricular septal defect (AVSD), ventricular septal defect
(VSD), atrial septal defect (ASD), and persistent ductus arteriosus
(PDA) in Down syndrome patients,
PDA, ASD, and VSD in Patau syndrome
patients, AVSD,
PDA and valvular defects in Edwards syndrome,
bicuspid aortic valve, aortic coarctation
and aortic stenosis in
Turner syndrome, and supravalvular
aortic stenosis and pulmonary stenosis in Williams syndrome.
Keywords: Heart defects
congenital; Down syndrome; Trisomy 13 syndrome; Trisomy 18 syndrome; Turner Syndrome; Williams Syndrome (source: MeSH NLM).
Introducción
Los síndromes cromosómicos hacen
referencia a cualquier alteración en el número o estructura normales de los
cromosomas, entre ellas tenemos a las aneuploidías, caracterizadas por la
pérdida o ganancia de material genético dando
como resultado un número anormal
de cromosomas, y las deleciones, que son defectos estructurales consecuencia de
la pérdida de un número variable de
genes. Estas alteraciones cromosómicas
predisponen a aquellas personas que los presentan a padecer enfermedades
congénitas, siendo las más comunes las cardiopatías congénitas (1).
Las malformaciones cardiacas, término
usado para definir las anormalidades en el corazón y sus grandes vasos,
consecuencia de un fallo en su embriogénesis, se detectan en 3-5% de los recién
nacidos y 1 de cada 33 cursa con una anomalía grave (1,2). Los avances
tecnológicos en la genética han permitido esclarecer mejor el rol de las
alteraciones cromosómicas en la génesis de las cardiopatías congénitas y su
asociación con algún síndrome.
La presente revisión se centrará en la
evidencia actual de las cardiopatías congénitas asociadas a síndromes
cromosómicos frecuentes, debido al rol trascendente que tiene la evolución
natural de este tipo de patologías en el desarrollo de los niños. Esto, con el
fin de dar al profesional de salud información relevante que apoye a la
sospecha diagnóstica en pacientes con sintomatología cardíaca y que presenten
un determinado tipo de síndrome, para que de esta manera se determine el
tratamiento
-clínico o quirúrgico- más adecuado, y
así mejorar la calidad de vida de la persona afectada. El objetivo de la
revisión es describir la frecuencia y analizar las características de las
principales cardiopatías
congénitas asociadas a síndromes cromosómicos
más prevalentes en pacientes
pediátricos.
Metodología
Se realizaron búsquedas en base de
datos electrónicas como MEDLINE, LILACS, SCIELO, y Google Scholar.
En el caso de MEDLINE se usaron términos MeSH y
términos libres, en tanto que para las otras bases de datos se recurrieron a
las palabras clave y uso de conectores con el fin de reducir los resultados a los más
trascendentes. Los resultados de la búsqueda final se limitaron a artículos en
inglés y español publicados
entre los años 2000 y 2021 en seres humanos; las
búsquedas iniciales se hicieron sin limitaciones para analizar si había algún
artículo en otro idioma, pero que sea importante.
La selección de artículos por evaluar
se realizó mediante la identificación de títulos y resúmenes que describían la
frecuencia de cardiopatías congénitas asociadas a pacientes con anomalías cromosómicas,
para lo cual se hizo una búsqueda individualizada por cada una de las anomalías
cromosómicas que se tratarán en la presente revisión (síndrome Down, síndrome
de trisomía 13, síndrome de trisomía 18, síndrome de Turner y síndrome de
Williams), por ejemplo: “Cardiopatías congénitas asociadas a síndrome Down”
(Figura 1).
Síndrome Down
Es la anomalía cromosómica más
frecuente, se da principalmente por tres mecanismos: trisomía 21 (95%),
translocación robertsoniana (3-4%) y mosaicismo de
trisomía 21 (1-2%) (3);
en casi el 90% de casos de trisomía 21, el cromosoma 21 extra se origina en la
madre, por tal motivo el riesgo aumenta
con la edad materna avanzada. Según la OMS, se estima que la prevalencia
global es de 10 en cada 10 000 recién nacidos vivos, pero se hace hincapié en
que las cifras dependen de variaciones socioculturales como la legalización del
aborto y el diagnóstico prenatal precoz (4).
Puede afectar a prácticamente todos
los sistemas y órganos, pero
entre las afecciones
más prevalentes se
hallan la dificultad para el
aprendizaje, hipotiroidismo, cardiopatías congénitas, alteraciones a nivel
gastrointestinal y leucemias. El diagnóstico es guiado por los criterios de
Hall, evaluados en todo recién nacido vivo y se confirma por citogenética (4).
Es la trisomía con mejor tasa de supervivencia, de hecho, un estudio de cohorte
retrospectiva evalúo 16 506 nacidos vivos entre los años 1982- 2003,
encontrando que las tasas de supervivencia al mes, 1, 5 y 20
años fueron de 98%, 93%, 91% y 88%,
respectivamente (5).
Se reportaron morbilidades asociadas
como hipertrofia adenoidea, asma, hernia umbilical, e hipotiroidismo (6), la
complicación más frecuente fue la
hipertensión pulmonar, casi el 50% lo padece y se presenta más en
asociación al defecto del tabique auriculoventricular; sin embargo, la
literatura enfatiza en que bebés no afectados inicialmente con hipertensión
pulmonar pueden volverse sintomáticos en la infancia o luego; asimismo, también
se presentaron insuficiencia mitral, infecciones respiratorias y edema pulmonar
(7).
Aproximadamente 50% de niños con
síndrome Down padece de algún tipo de cardiopatía congénita y la mayor
mortalidad se halla en los dos primeros años (8). En un estudio retrospectivo
realizado en Marruecos se calculó que la tasa de mortalidad fue 14,1% (8), esto
nos muestra un gran contraste con Suecia, en donde la tasa de mortalidad ha
disminuido de 41 a 4 % en un período de
50 años desde 1973 hasta 2003 (9); en Latinoamérica, Panamá, reporta una
mortalidad de 3,45% (6).
Además,
un gran estudio poblacional en
Inglaterra halló que la mortalidad se reducía de 30 a 5% luego
de una intervención quirúrgica temprana (10). Las principales causas de mortalidad
son choque séptico y cardiogénico (11,12), por lo que se justifica que los
pacientes con Síndrome Down tengan investigación en el período neonatal y
postnatal, con la finalidad de reducir la morbilidad y mortalidad en relación
con las complicaciones (13,14). En la Tabla 1 se describen las principales
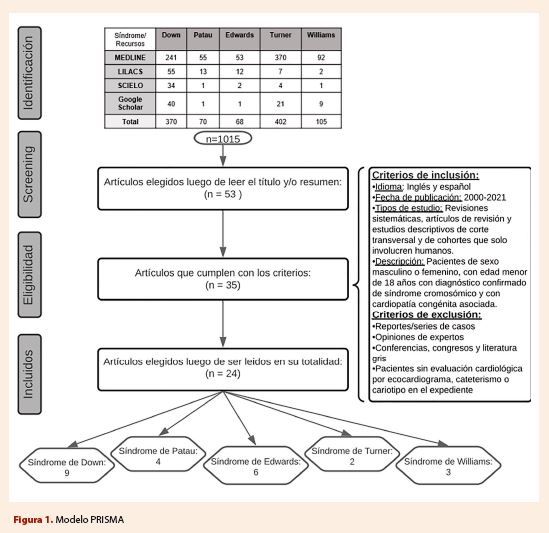
cardiopatías congénitas frecuentemente asociadas con el síndrome Down.
Síndrome
Patau
Este síndrome cursa con tres
mecanismos patogénicos: trisomía 13, translocación robertsoniana
y mosaicismo. La tríada clínica característica se constituye por micro/anoftalmia, paladar
hendido y polidactilia posaxial (15). Generalmente padecen de múltiples
alteraciones ya sean anatómicas o funcionales, siendo el riesgo de muerte fetal
del 80%, pues se asocian malformaciones craneofaciales, oculares, cerebrales,
hematológicas, abdominales
y cardiopatías, debido a esto el pronóstico no es muy bueno,
siendo la supervivencia menor a 1 año en su mayoría y sobrepasar los 10 años es
excepcional (16). La prevalencia es variable entre 1 de 10 000 nacidos vivos o
1 de 30 000 nacidos vivos. Es la tercera trisomía en frecuencia luego de la
trisomía 21 y 18, dependiente de factores socioculturales (16).
Las cardiopatías de la trisomía 13 con
frecuencia son múltiples, es raro que se hallen aisladas y que solo una
cardiopatía congénita afecte al paciente pediátrico. El diagnóstico se sospecha
con ecografía del primer trimestre de embarazo másel
uso de marcadores cromosómicos, y en el segundo trimestre con estudio
morfológico, en tanto, la confirmación se da con el cariotipo (16). Las
cardiopatías congénitas se presentan en el 80% de casos, la mediana de
supervivencia es de 7 días y la mayoría fallece en el primer mes de vida (17). En un estudio se halló que la menor frecuencia de anomalías
cardíacas contribuye a una supervivencia más prolongada (18).
La principal causa de óbito son las
complicaciones cardiopulmonares, el 50% fallece el primer mes de nacido y el
70% a los 6 meses; sin embargo, la mortalidad intrahospitalaria disminuye en
45% en pacientes que se someten a intervención quirúrgica (19). Por ello, si
bien la mayoría de
pacientes fallecen en las primeras
semanas luego del nacimiento, algunos pueden sobrevivir más allá del primer año
(20,21). En la literatura revisada se encontraron varias cardiopatías
congénitas frecuentemente asociadas con síndrome Patau
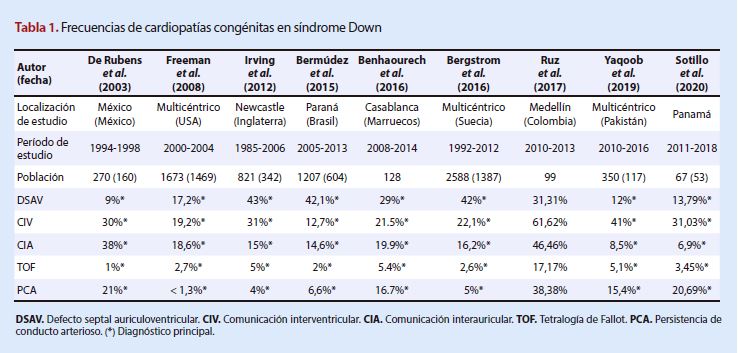
(Tabla 2).
Síndrome Edwards
Es un desorden genético causado por la
presencia, ya sea total o parcial, de un cromosoma 18 adicional, este síndrome
es la segunda trisomía más frecuente después del síndrome Down. Su prevalencia
global se encuentra entre 1 de 7000 nacidos vivos, la cual aumenta conforme la
edad materna sea mayor. La tasa de mortalidad después del nacimiento es elevada,
con un porcentaje del 50% que sobrevive después de la primera semana y un 5-10%
que logra superar el año de edad (22). El diagnóstico de estos pacientes se
realiza, en su mayoría, durante la etapa prenatal por la presencia de
malformaciones anatómicas en el ultrasonido (translucencia nucal, retardo en el
crecimiento) o por el cariotipo después de una amniocentesis o cordocentesis
(22).
Las manifestaciones clínicas de este
síndrome incluyen: alteraciones en el crecimiento, retraso psicomotor, cardiopatías
congénitas, anomalías faciales, malformaciones del cráneo, el tórax, el abdomen
y genitales (23). Siendo los defectos cardíacos congénitos las manifestaciones
clínicas más frecuentes (90% de los casos), además de ser la principal causa de
muerte (24). Entre las más frecuentes tenemos: defectos del septo, ductus
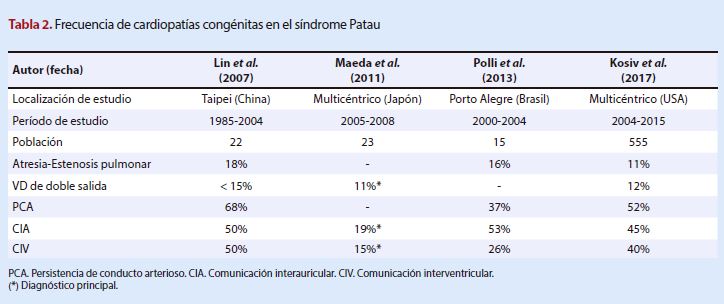
arterioso persistente y defectos valvulares (19) (Tabla 3).
Síndrome Turner
También llamado síndrome de Ullrich-Turner o monosomía X; como su propio nombre lo
indica, es causado por la ausencia parcial o total del segundo cromosoma X
siendo la única monosomía compatible con la vida. La gran mayoría de
gestaciones «45, X» concluyen en abortos espontáneos, teniendo como principal
causa las anormalidades cardíacas (25,26). Se estima que el síndrome Turner
está presente en el 3% de fetos femeninos, de los cuales un 10% sobrevive;
asimismo, tiene una prevalencia de 1 de 2500 nacidas vivas. No se encontraron
datos de mortalidad posnatal, salvo por un estudio prospectivo realizado con
156 mujeres en 1986, seguidas por 17 años, con 15 de ellas fallecidas en este
transcurso (3,6%) (27).
El diagnóstico se
realiza mediante la
identificación de características
clínicas reconocibles y el cariotipo (26). Este síndrome se caracteriza por la
baja estatura, retraso intelectual, falla ovulatoria prematura, anomalías
faciales, cuello alado, linfedema y defectos cardíacos congénitos. El fenotipo
que pueden presentar es muy variable entre cada paciente.
El porcentaje de mujeres que presentan
cardiopatías congénitas varía según distintas bibliografías (25-50%) siendo las
más frecuentes aquellas que afectan al corazón izquierdo: la válvula aórtica
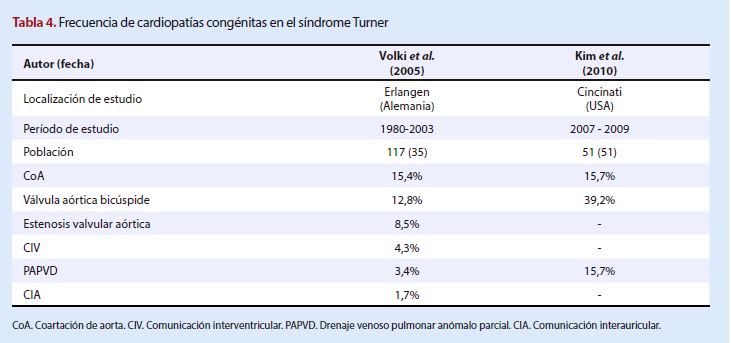
bicúspide, coartación de aorta y estenosis valvular aórtica (26,27). En la
Tabla 4 se detalla la literatura revisada con las principales cardiopatías
congénitas asociadas al síndrome Turner.
Síndrome Williams
Este
síndrome también
conocido como síndrome de
Williams- Beuren, es causado por la pérdida de 26 a
28 genes debido a la deleción del cromosoma 7, específicamente 7q11.23. La
prevalencia de esta afección se encuentra en 1 de 10 000 nacidos vivos.
Producto de esta anormalidad genética se presentan alteraciones que involucran
distintos sistemas, entre ellos: nervioso, hematológico, tejido conectivo,
osteoarticular, cardiovascular, entre otros (28). El compromiso cardiovascular
está dado por la afectación del gen que codifica a la elastina (ELN) lo que
conlleva a un depósito
anormal o deficiente de esta
proteína durante el
periodo embrionario, muy
importante para la formación de los vasos sanguíneos. Las malformaciones
congénitas que atacan
al sistema cardiovascular están presentes en cerca de un 75-80%
de los pacientes (29).
En
la bibliografía revisada, las
alteraciones vasculares
como la estenosis de la arteria pulmonar y la estenosis aórtica supravalvar fueron los defectos más frecuentes
a comparación de las alteraciones intracardiacas que eran
más catalogadas como hallazgos atípicos (28-30). En la literatura revisada se
encontraron varias cardiopatías congénitas asociadas al síndrome Williams
(Tabla
5).
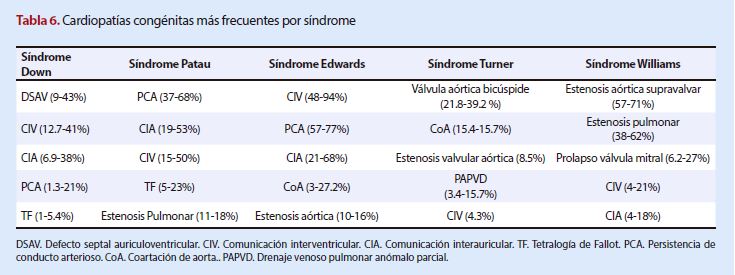
En
resumen, y según lo revisado en la literatura, las cardiopatías congénitas más
frecuentes asociadas a síndromes cromosómicos se describen en la Tabla 6.
Discusión
Síndrome Down
Si
bien la cardiopatía congénita más asociada al síndrome Down fue el defecto en
el tabique AV, hubo gran variabilidad en el
tamaño
de las muestras y el porcentaje obtenido, siendo los más elevados en
Inglaterra, Brasil y Suecia, en contraste con Panamá, Pakistán y México, el
cual según su población estudiada solo estuvo presente en 6% de la muestra.
La comunicación interventricular fue más frecuente en Colombia y Pakistán, en donde el
62 y 41% de sus muestras padecían este tipo de cardiopatía congénita, en las
demás poblaciones de estudio no se halló mayor variación, y la frecuencia se
hallaba entre 13-31%. Colombia también obtuvo los mayores índices de
comunicación interauricular.
La tetralogía de Fallot fue una
cardiopatía bastante infrecuente, los estudios obtuvieron una variación en el
rango de 1-5,5%, a excepción de Colombia en donde llegó a 17,17%. Por otro lado, la persistencia del conducto
arterioso fue más frecuente en Colombia, México, Panamá, Marruecos y Pakistán;
los otros estudios descriptivos de países como Estados Unidos, Inglaterra,
Suecia y Brasil tuvieron una frecuencia de entre 1-7%.
La explicación sobre la gran
variabilidad de datos expresados como porcentajes son la variación
sociocultural y la mayor capacidad de diagnóstico de cardiopatías congénitas
con el paso de los años; asimismo, la gran proporción en el estudio de Colombia
puede hallar su explicación parcial en que sus porcentajes no excluyeron datos
de cardiopatías múltiples, a diferencia de los demás estudios, en donde se
consideró solo el recuento de cardiopatías aisladas.
Si bien las cardiopatías congénitas se
diagnostican hasta en 50% de pacientes con síndrome Down, la frecuencia es
menor en aquellos con mosaicismo por trisomía 21 (1). El hecho es que los
defectos cardiacos congénitos se han vuelto menos comunes en bebés
diagnosticados con síndrome Down, probablemente por mejoras en el diagnóstico
prenatal y aborto selectivo (9).
Síndrome Patau
A pesar de los pocos estudios
obtenidos y de la muestra reducida de estos, la cardiopatía más asociada en
China y Estados Unidos fue la persistencia del ductus arterioso, a diferencia
de Brasil y Japón, en donde los primeros lugares lo ocupan los defectos en el tabique interauricular e
interventricular. La atresia-estenosis pulmonar tuvo un rango de entre 11-18%,
pero en el estudio de Japón la frecuencia fue nula.
La tetralogía de Fallot fue más
frecuente en China en comparación con los demás estudios descriptivos; por otro
lado, el ventrículo derecho de doble salida y la coartación de aorta obtuvieron
frecuencias similares en China, Japón y Estados Unidos, cabe reiterar que en
Brasil no se obtuvieron datos sobre estas cardiopatías.
Se comparte la explicación brindada
para el síndrome Down, además, es importante detallar que en el estudio de
Japón se consideraron porcentajes de cardiopatías congénitas aisladas y no en
forma de múltiples como en los estudios de China, Brasil y Estados Unidos.
Síndrome Edwards
Se presenta a la comunicación
interventricular como la cardiopatía congénita más frecuente (48-94%) en todos
los estudios revisados, siendo también la más frecuente reportada por las
bibliografías revisadas (2) donde se la presenta con una frecuencia mayor al
90%, seguido por la PCA, con muchas más variaciones entre cada uno de los
estudios.
Se revisaron estudios que solo
consideraban dentro de la estadística a los diagnósticos principales de cada
caso, lo cual explicaría el porqué de variaciones muy pronunciadas en los
porcentajes, además de subestimar la frecuencia de otras patologías que podrían
estar presentes concomitantemente en cada uno de los pacientes evaluados. Esto
dificulta la comparación de los datos y conocer los valores absolutos de cada
una de ellas.
Existieron estudios que no reportaron
la presencia de algunas cardiopatías, como en el caso de estenosis aórtica,
insuficiencia aórtica y atresia de la válvula aórtica, lo que puede ser
explicado por la diferencia en el número de poblaciones evaluadas, puesto que
algunos estudios presentaban 22 casos revisados frente a otro con 1020
pacientes estudiados.
Síndrome Turner
Las dos cardiopatías congénitas más
frecuentes en el síndrome Turner observadas en los dos estudios revisados
fueron aquellasque comprometen a la arteria y válvula
aórticas: coartación de aorta y válvula aórtica bicúspide. Esto se correlaciona
con lo descrito por Bondy (27) que hizo una revisión a cinco estudios con más
de cien pacientes, con fechas de publicación anteriores a nuestra búsqueda, en
los cuales se determinó que la válvula aórtica bicúspide (12-30%) y la
coartación de aorta (7-18%) son las presentaciones más frecuentes, seguidas por
defectos septales y circulación pulmonar venosa anómala parcial. Sin embargo,
Lin (2) describe otras anomalías que presentan una frecuencia similar a las
mencionadas: elongación de aorta transversal (25-50%), dilatación aórtica
(25-40%) y anomalías coronarias congénitas (5-25%).
La frecuencia de coartación de aorta coincide en los estudios que
se analizaron (26,27), pero no hubo una correspondencia en los porcentajes de
las demás cardiopatías e, incluso, no se
reportó la presencia de otras como la estenosis
valvular aórtica o las comunicaciones intercavidades.
La poca cantidad de estudios
encontrados y la diferencia entre el número de población no permitiríanunabuenacomparaciónentrelosresultadosencontrados.
Esto agregado a que no se reportaron otras cardiopatías congénitas que sí
fueron consideradas en el estudio de Volkl (25).
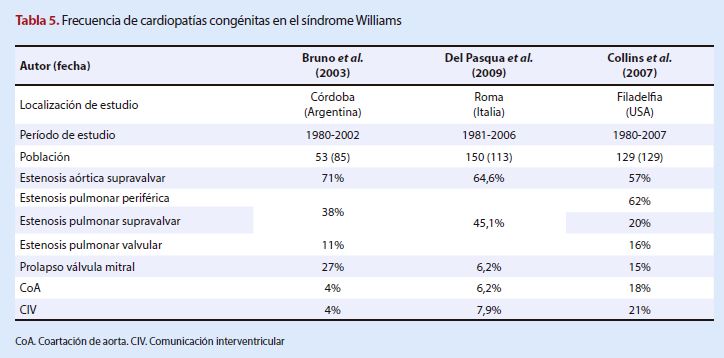
Síndrome Williams
Los tres estudios revisados señalan a
la estenosis aórtica supravalvar como la
manifestación anómala cardíaca más frecuente en este síndrome. La literatura
científica también la presenta como la principal, con frecuencias que van desde
45 al 75% (28). Lo mismo sucede con la estenosis pulmonar en sus diferentes
presentaciones (periférica, supravalvar, valvular)
con frecuencias que la sitúan en un rango de 37-75%, con la mayoría de estudios
que reportan una frecuencia del 40% (29).
El prolapso de válvula mitral
representa el 27% de casos en Argentina, frecuencia un poco mayor
en comparación con los Estados Unidos de Norteamérica (EUA)
(15%) e Italia (6,2%). La coartación de
aorta y la comunicación
interventricular tuvieron similar frecuencia en Argentina e
Italia, pero en EUA el porcentaje aumentó. Por todo lo mencionado se puede inferir que las
poblaciones de Argentina e Italia son similares en cuanto a cardiopatías congénitas asociadas a
síndrome de Williams, y en su mayoría los porcentajes se elevan en el caso de
EUA, en donde es más frecuente la estenosis pulmonar periférica.
Limitaciones
Los
estudios revisados, en su mayoría, tienen fechas de publicación de la década
pasada lo cual dificulta conocer la actualidad de estas cardiopatías. Existe
una cantidad limitada de estudios que tratan sobre cardiopatías asociadas a
síndromes cromosómicos diferentes al síndrome Down; además, contar
con un número de población reducida no permite tener
una aproximación tan exacta a la frecuencia real de estas patologías. La nacionalidad, los tipos de estudios
(algunos multicéntricos), metodología, la forma de presentación de la
información y resultados son muy variados, esto dificulta la posibilidad de
realizar una comparación y posterior análisis de los mismos. Finalmente, la
ausencia de publicación de artículos nacionales de este tipo no permite conocer
información relevante sobre la situación actual del país, por ello la necesidad
de realizar investigación científica sobre estos temas.
Conclusiones
La
frecuencia de cardiopatías congénitas en niños con síndromes cromosómicos es
alta, pero muy variable, por tal motivo, se recalca la importancia de
investigación en el período neonatal, para así disminuir las tasas de
morbilidad y mortalidad, y asegurar una mejor calidad de vida en el desarrollo
de estos niños.
Contribución de los
autores
JECL
y OEEA: diseño de estudio, extracción y análisis de datos, redacción del
manuscrito. GPHB: diseño de estudio, revisión del manuscrito y aprobación de la
versión final del manuscrito.
Referencias
bibliográficas
1.
Zaidi S, Brueckner M. Genetics and Genomics of Congenital
Heart Disease.
Circulation Research.
2017;120(6):923-940. DOI: 10.1161/
CIRCRESAHA.116.309140
2.
Lin A, Santoro S, High F, Goldenberg P, Gutmark‐Little I. Congenital heart defects associated
with aneuploidy syndromes: New insights into familiar
associations.
American Journal of Medical
Genetics Part C: Seminars in Medical
Genetics. 2019;184(1):53-63. DOI: 10.1002/
ajmg.c.31760
3.
Jones KL. Smith’s recognizable patterns of human malformation, 6th
ed, Elsevier Saunders, Philadelphia 2006.
4.
Díaz Cuéllar S, Yokoyama
Rebollar E, Del Castillo Ruiz V. Genómica
del síndrome de Down. 2016:289-295.
5.
Kucik JE, Shin M, Siffel C, Marengo L,
Correa A. Trends in survival
among children with Down syndrome in 10 regions of the
United States. Pediatrics 2013;
131:e27. DOI: 10.1542/peds.2012-1616
6.
Sotillo-Lindo
J, Barrantes I. Prevalencia y perfil
de cardiopatías congénitas en pacientes con
Síndrome de Down. Hospital de Especialidades Pediátricas Omar
Torrijos Herrera. 2011-2018. Revista Pediátrica de Panamá. 2020;37-40. DOI: 10.37980/im.journal. rspp.20201695
7.
Ruz-Montes M, Cañas-Arenas E, Lugo-Posada M, Mejía-Carmona M, Zapata-Arismendy
M, Ortiz-Suárez L, et al.
Cardiopatías congénitas más frecuentes en niños con síndrome de Down. Revista
Colombiana de Cardiología.
2017;24(1):66-70. DOI: https://doi.org/10.1016/j. rccar.2016.06.014