Reporte de Caso
Taquicardia ventricular
polimórfica catecolaminérgica en adolescente: un diagnóstico clínico, electrocardiográfico
y genético
Catecholaminergic polymorphic
ventricular tachycardia in adolescents:
a clinical, electrocardiographic
and genetic diagnosis
María
Concepción Rocha-Arrieta1,a,
https://orcid.org/0000-0003-0823-6918
Antonio
Arias-Díaz2,b,
Carlos
Alberto Quiróz-Romero3,c,
Yermis
Rocha-Arrieta4,d
https://orcid.org/0000-0002-0390-8255
DOI:
https://doi.org/10.47487/apcyccv.v2i3.151
Resumen
La taquicardia ventricular polimórfica
catecolaminérgica es una de las canalopatías más
letales. Los síntomas de la enfermedad aparecen en la niñez o la adolescencia,
los cuales están caracterizados por arritmias ventriculares desencadenadas por
estrés o actividad física. Se presenta el caso de una adolescente que consultó
por síncopes recurrentes precipitados por el ejercicio. En el abordaje
diagnóstico se determinó como taquicardia ventricular polimórfica catecolaminérgica,
con mutación en el gen del receptor de la rianodina cardíaco, heterocigoto
c.14311G>A(p.v4771I exón 100), para el manejo fue
necesario antiarrítmicos y el implante de un cardiodesfibrilador,
con evolución satisfactoria. La sospecha clínica, la prueba de esfuerzo y las
pruebas genéticas son fundamentales para un diagnóstico y manejo oportuno de esta
patología.
Palabras clave: Taquicardia ventricular;
Síncope; Muerte súbita; Genética; Rianodina (fuente: DeCS
BIREME).
Abstract
Catecholaminergic polymorphic ventricular tachycardia
is one of
the most lethal channelopathies, characterized by ventricular arrhythmias triggered by stress or physical
activity. We present
the case of an adolescent
who consulted for recurrent syncope
precipitated by exercise. In the diagnostic approach, catecholaminergic polymorphic
ventricular tachycardia was
reached, with a mutation in the cardiac ryanodine
receptor gene, Heterozygous c.14311G> A (p.v4771I exon 100), antiarrhythmic drugs and implantable cardioverter-defibrillator
were necessary with satisfactory evolution. Clinical suspicion, stress test and genetic
tests are essential for a timely diagnosis and management of this
pathology.
Keywords: Tachycardia,
ventricular; Syncope; Death,
sudden; Genetic; Ryanodine (source: MeSH NLM).
Introducción
La taquicardia ventricular polimórfica
catecolaminérgica (TVPC)
es una canalopatía heredable,
caracterizada por la aparición
de contracciones ventriculares monomórficas progresando a taquicardia ventricular
bidireccional y/o polimórfica durante el esfuerzo físico, estrés emocional o perfusión de catecolaminas
en corazones estructuralmente íntegros, sin alteraciones en el
electrocardiograma (ECG) basal. La prevalencia estimada de TVPC es de 1-5 por 10
000 individuos y cerca del 30% de los pacientes tiene una historia familiar de muerte
cardiaca súbita (1,2).
Generalmente, las manifestaciones clínicas
debutan entre los 6 – 10 años de edad (2) y las más comunes son: el síncope
recurrente (80%) desencadenado durante la actividad física, parada cardiorrespiratoria
(30%) y, en el peor de los casos, muerte súbita que en niños y adolescentes es infrecuente,
aunque en adultos jóvenes sin tratamiento se han registrado en el 30% de los casos
(3,4).
Con las investigaciones recientes se han podido categorizar múltiples causas de la TVCP,
como son: 1) TVPC con mutaciones o juveniles: aproximadamente el 55% de los
pacientes presentan mutaciones en el gen RyR2, receptor cardíaco de rianodina tipo
2; localizado en el brazo largo del cromosoma 1 (1q43), con un patrón de herencia
autosómica dominante (5). Mutaciones en el gen de la calsecuestrina
cardíaca 2 CASQ2 (1p13.1) con un patrón autosómico recesivo presente en el 2% de
los casos (2). También se han descrito otras mutaciones con una incidencia
menor en los genes de la triandina, trianina y adenilato quinasa 2 (5); 2) TVPC esporádica o no
genotipificada, representa el 39% de los casos,
afectando principalmente a mujeres mayores de 20 años (5), y 3) la TVPC relacionada
con otras patologías como el síndrome de Andersen-Tawil,
y el síndrome de QT Largo tipo 4 (5).
Para el tratamiento de la TVPC, los betabloqueadores son los más usados, a pesar de su efectividad
limitada. También se ha propuesto el uso combinado de betabloqueadores,
antagonistas de calcio y flecainida; además, la
denervación simpática y cardiodesfibrilador
implantable (CDI)(6). Finalmente, en todos los pacientes se debe evitar
actividades de alto impacto y situaciones de estrés (6).
A continuación, se presenta el caso
clínico de una paciente con TVPC, este reporte tiene como objetivo llamar la
atención del clínico ante un síncope cardiogénico donde la TVPC se debe
mantener bajo sospecha; asimismo, resaltar la importancia de la biología
molecular en el abordaje diagnóstico de las arritmias en pediatría.
Reporte del caso
Paciente femenina de 11 años de edad
que presentó síncope jugando fútbol, luego de 3 meses repite el episodio mientras
corría. No tenía antecedentes personales patológicos ni de
enfermedad cardíaca tampoco de muerte
súbita en la familia. Durante el interrogatorio la paciente
manifestaba que ante emociones fuertes presenta visión
borrosa y náuseas. En el examen físico el peso y talla fueron adecuados
para la edad, sin alteración cardiovascular. Los padres negaron cambios en la
dinámica familiar o en el comportamiento
de la menor.
Se indicó la prueba de mesa basculante
con resultado positivo para síncope neurocardiogénico
tipo vasodepresor por lo que se consideró como diagnóstico
inicial de síncope vasovagal, manejado con aumento en
el consumo de sal luego de los eventos.
A los 13 años de edad la paciente
presentó episodios presíncopes recurrentes desencadenados
durante el ejercicio, por lo cual se decide realizar un electrocardiograma Holter
de 24 h, que reporta ritmo sinusal de base durante la mayor parte del estudio, con
extrasístoles ventriculares multifocales ocasionales (Figura 1).
Se recomendó evitar situaciones de estrés
físico y emocional, simultáneamente se instaura medicación con metoprolol 1,5 mg/kg/día. A los 2 meses de iniciado
el betabloqueador mientras la paciente nadaba presentó
perdida de conciencia acompañado de cianosis peribucal durante 3 min, por lo que
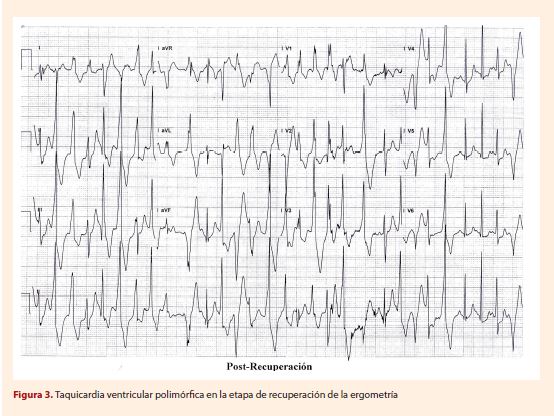
se solicita ecocardiograma transtorácico sin anormalidades, con buena función biventricular. También se le realizó la prueba de esfuerzo modificada
en la que alcanzó el 93% de la frecuencia cardiaca máxima, observándose extrasístoles
ventriculares desde la etapa 2, inicialmente monomórficas sugestivas de originarse
en el tracto de salida del ventrículo derecho, posteriormente polimorfas y alternantes
en su polaridad, progresando en etapa 3 a evento de taquicardia ventricular polimorfa
no sostenida hasta ocho latidos. Al suspender la prueba, presenta rápida recuperación
de ritmo sinusal (Figura 2 y 3). Adicionalmente, se realizó resonancia
magnética nuclear contrastada que descartó displasia arritmogénica
ventricular.
Finalmente, a la paciente, padres y hermanos
se les realizó un panel molecular para TVPC, donde solo se detectó en la paciente
una mutación en el gen RyR 2 (heterocigoto c.14311G>A(p.V4771I)).
En junta médico-quirúrgica se decide
llevar a ablación con mapeo tridimensional con catéter array en tracto de salida
del ventrículo derecho, durante este se induce fácilmente a taquicardia ventricular
polimórfica con diferentes extrasístoles que la gatillan. Con este tratamiento se
logra modular parcialmente las zonas iniciales de descarga, pero persiste con inducibilidad de focos, que sugieren origen en otras zonas
del ventrículo derecho o regiones septales izquierdas
Ante la mala respuesta a las medidas iniciales,
se consideró que la paciente era candidata a implante de cardiodesfibrilador.

El procedimiento se realizó sin complicaciones
y se adicionó a la terapia propafenona 150
mg cada 12 h. Después de 8 años del procedimiento la paciente requirió tres hospitalizaciones, en la última
se cambia el tratamiento a amiodarona y carvedilol, lo que disminuyó
considerablemente los eventos, no obstante, la paciente manifiesta en la
actualidad incomodidad ante las descargas y le angustia la posibilidad que el
dispositivo no funcione; sin embargo, tiene buena adherencia al tratamiento y a
las medidas no farmacológicas.

Discusión
Entre las canalopatías
primarias, la TVPC es una de las más letales con un impacto social importante por
el grupo etario afectado (7). La manifestación clínica en este caso fue el síncope
recurrente; las guías recomiendan el ECG como primer estudio en síncope, aunque
en TVPC es un dilema por la normalidad en el ritmo de base (6). Algunas series de
casos informan que hasta el 60% de los pacientes son manejados con otros
diagnósticos relacionados con causas vasovagales y neurológicas
lo que causa un retraso en el diagnóstico de TVPC hasta de 2 años (8,9), como sucedió
en el presente reporte, donde el estudio inicial fue la prueba de mesa
basculante; recientemente, el uso de esta herramienta en niños ha presentado controversias
por la variabilidad de interpretación; además, un resultado positivo no descarta
causas potencialmente deletéreas y la toma de decisiones basadas en ello no está
exenta de riesgos para el paciente (10).
En este caso, la TVPC se consideró con
el Holter de 24 h, el diagnóstico de certeza se establece con la prueba de esfuerzo
(6) donde se pudo apreciar la progresión de latidos ectópicos ventriculares
hasta taquicardia polimórfica, por lo que se realizó la prueba genética (6).
La mutación descrita en este reporte fue en el gen
RyR2, que es un gran canal iónico con peso de 546kD de forma homotetramérica, en el cardiomiocito está localizado en retículo
sarcoplásmico (RS), su función es regular la liberación de calcio (Ca2+) desde el
lumen del RS hacia el citoplasma durante la contracción cardíaca (11). Actualmente,
las mutaciones registradas de RyR2 con cambio de sentido (missense)
son alrededor de 150,
muchas de estas asociadas al desarrollo de TVPC (12); las mutaciones se distribuyen
en tres puntos calientes llamados dominios, agrupando un número de aminoácidos:
N terminal (164–433), dominio central (2,246–2,504) y C-terminal (3,778–4,959) (13).
En este reporte la variante fue c.14311G>A(p.v4771I) que es una mutación missense
considerada patógena, y que ha sido reportada en asociación con TVPC (14), se encuentra
localizada en la región C-terminal que conforma el poro del canal. En esta
mutación se da un cambio de una valina por isoleucina, si bien el cambio es de
un aminoácido no polar por otro no polar y supone pequeñas modificaciones en las propiedades
fisicoquímicas de hidrofobicidad y masa, este residuo de valina es altamente
conservado entre las especies y está localizado en uno de los tres puntos calientes para mutaciones con
potencial fisiológico, por ende, se asume un rol importante de esta mutación en
la ganancia de función de la RyR2 (13,15).
Existen varias hipótesis del porqué las
mutaciones en RyR2 permiten la TVPC, la mayormente aceptada es que las
mutaciones sensibilizan los canales de RyR2 al calcio luminal del retículo
sarcoplásmico permitiendo su apertura a una concentración más baja de calcio intracelular,
lo que se ha denominado liberación de calcio inducida por sobrecarga de almacenamiento
(SOICR, del inglés store overload-induced calcium release). Según esta hipótesis,
al disminuir el umbral de activación se permite fugas de calcio durante la diástole
lo cual precipita las arritmias (11). Otra teoría más reciente sugiere que las mutaciones
alteran la interacción interdominios desestabilizando
el cierre del canal y permitiendo fugas del Ca2+ contenido en el RS (11,13).
Con
respecto al tratamiento
de la TVPC, el
manejo inicial en la paciente fue reducir actividades de alto impacto y
la administración de betabloqueadores, primera línea de manejo para TVPC (6), pero no son suficientes
para controlar los eventos, por lo que se lleva a ablación con catéter array, dicho procedimiento
es considerado como terapia adyuvante en cuadros refractarios, aunque hay pocos
reportes de éxito (6,7), tal y como se evidenció en esta paciente. Luego de la falla
terapéutica se decide el implante de CDI, lo cual ha sido recomendado en las guías
(6,7); no obstante, se ha registrado que el 50% de los niños tienen descargas
inadecuadas lo que genera dolor, ansiedad y depresión aumentando el riesgo de
arritmias (16). La propafenona, al igual que la flecainida, han logrado demostrar la inhibición de actividad
RyR2 con resultados clínicos a largo plazo favorables, aunque los recientes estudios
cuestionan el mecanismo de acción de la flecainida sobre
el RyR2 (7); en algunas situaciones de recurrencia es necesario el uso de combinaciones
de antiarrítmicos, como en este caso (17). Pese a todos los efectos secundarios
del dispositivo, los padres y la paciente consideran que ante el eventual riesgo
de muerte súbita los beneficios del tratamiento superan el riesgo. Finalmente, las
investigaciones recientes aplicando la tecnología de edición de genes CRISPR/Cas9
parece ser prometedor en esta patología (12).
Conclusión
Considerar la TVPC en los niños con síncopes
inducido por emociones o actividad física es importante por la gravedad de esta
condición; en países como Colombia contar con herramientas de la biología molecular
es esencial para el diagnóstico e intervención terapéutica adecuada, así como la
identificación oportuna en los familiares de los pacientes con TVPC, permitiendo
un diagnóstico presintomático y consejería genética.
Referencias
bibliográficas
1.
Schwartz P, Ackerman M, Wilde A. (2017). Channelopathies as Causes
of Sudden Cardiac Death. Cardiac Electrophysiology Clinics, 9(4), 537-549.
https://doi.org/10.1016/j.ccep.2017.07.005
2.
Pflaumer A, Davis AM. Guidelines for
the diagnosis and management of Catecholaminergic
Polymorphic Ventricular Tachycardia.
Heart Lung Circ. 2012 Feb;21(2):96-100. doi:
10.1016/j.hlc.2011.10.008. Epub 2011 Nov 25. PMID: 22119737.
3.
Roston TM,
Cunningham TC, Sanatani S. Advances
in the diagnosis and
treatment of catecholaminergic polymorphic ventricular tachycardia. Cardiol Young. 2017 Jan;27(S1):S49-S56.
doi: 10.1017/
S1047951116002237. PMID: 28084961.
4.
Seidlmayer
LK, Riediger F, Pagonas N, Nordbeck P, Ritter
O, Sasko
B. Description of a novel RyR2
mutation in a juvenile patient with
symptomatic catecholaminergic
polymorphic ventricular
tachycardia in sleep
and during exercise: a case
report. J Med Case Rep.
2018 Oct 9;12(1):298. doi:
10.1186/s13256-018-1825-6. PMID: 30296944; PMCID: PMC6176516.
5.
Pérez-Riera AR, Barbosa-Barros R, de Rezende Barbosa MPC, Daminello- Raimundo R, de Lucca
AA Jr, de Abreu LC.
Catecholaminergic polymorphic
ventricular tachycardia, an
update. Ann Noninvasive Electrocardiol. 2018 Jul;23(4):e12512. doi: 10.1111/anec.12512. Epub 2017 Oct 19. PMID:
29048771; PMCID: PMC6931575.
6.
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al . HRS/ EHRA/APHRS expert consensus statement on the
diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May
2013 and by ACCF,
AHA, PACES, and AEPC in June
2013. Heart Rhythm. 2013 Dec;10(12):1932-63. doi:
10.1016/j.hrthm.2013.05.014. Epub 2013 Aug 30. PMID: 24011539.
7.
Sumitomo N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J Arrhythm. 2016 Oct;32(5):344-351. doi: 10.1016/j.joa.2015.09.008. Epub
2015 Nov 24. PMID: 27761157; PMCID: PMC5063269.
8.
Roston TM, Vinocur JM, Maginot KR, Mohammed S, Salerno JC, Etheridge SP, et al
. Catecholaminergic
polymorphic ventricular tachycardia in children: analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol. 2015 Jun;8(3):633-42.
doi: 10.1161/
CIRCEP.114.002217. Epub 2015 Feb 24. PMID:
25713214; PMCID: PMC4472494.
9.
Kawata H, Ohno S, Aiba T, Sakaguchi H, Miyazaki A, Sumitomo N,
et al.
Catecholaminergic polymorphic
ventricular tachycardia (CPVT) associated with ryanodine receptor (RyR2) gene mutations—long-term prognosis after initiation of
medical treatment. Circ J 2016;80:1907–15
10.
Sanatani S, Chau V, Fournier A, Dixon A, Blondin
R, Sheldon RS. Canadian Cardiovascular Society and Canadian Pediatric Cardiology Association Position Statement on the Approach
to Syncope in the
Pediatric Patient. Can J Cardiol. 2017 Feb;33(2):189-198. doi: 10.1016/j.cjca.2016.09.006. Epub
2016 Oct 3. PMID: 27838109.
11.
Wleklinski MJ, Kannankeril PJ, Knollmann BC. Molecular and
tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia.
J Physiol. 2020 Jul;598(14):2817-2834.
doi: 10.1113/ JP276757. Epub 2020 Apr 27. PMID:
32115705; PMCID: PMC7699301
12.
Wei H, Zhang XH, Clift C, Yamaguchi N, Morad M. CRISPR/Cas9 Gene editing of RyR2 in human
stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca2+ signaling. Cell Calcium. 2018 Jul;73:104-111. doi:
10.1016/j.ceca.2018.04.009. Epub 2018 Apr 27. PMID: 29730419; PMCID: PMC5993620.
13.
Jiang D, Jones PP, Davis DR, Gow
R, Green MS, Birnie DH, et al. Characterization of
a novel mutation
in the
cardiac ryanodine receptor that results in catecholaminergic polymorphic
ventricular tachycardia. Channels(Austin). 2010 Jul-Aug;4(4):302-10. doi: 10.4161/chan.4.4.12666. Epub 2010 Jul 14. PMID: 20676041; PMCID: PMC3322479.
14.
National Center for Biotechnology Information. ClinVar; [VCV000201357.5], https://www.ncbi.nlm.nih.gov/clinvar/variation/ VCV000201357.5
15.
Pérez
Ruescas C. Estudio
genético de muerte súbita en la Región
de Murcia [ Tesis Doctoral].
España: Universidad de Murcia:2015. Recuperado a partir
de: https://digitum.um.es/digitum/ handle/10201/45837
16.
Sumitomo N. Devide therapy in children and patients with congenital heart disease. J Arrhythmia. 2014;30:428-432.
17.
Viskin S, Chorin E, Viskin D, Hochstadt A, Schwartz AL, Rosso R. Polymorphic
Ventricular Tachycardia: Terminology,
Mechanism, Diagnosis,
and Emergency Therapy. Circulation. 2021 Sep
7;144(10):823-839. doi:
10.1161/CIRCULATIONAHA.121.055783. Epub 2021 Sep 7. PMID: 34491774
Filiación de los autores
1
Universidad de Cartagena. Carta- gena, Colombia.
2
Unidad de Cuidados Intensivos Pediátricos, Fundación
Unidad de Cuidados Intensivos Dona Pilar.
3 Fundación
Cardiovascular del Oriente Colombiano. Bucaraman- ga, Colombia.
4
Universidad de Antioquia. Mede-
a Médico, especialista en pediatría.
b
Cardiólogo pediatra.
c
Médico internista. Cardiólogo electrofisiólogo.
d
Microbióloga bioanalista, MsC.
Recibido:
27 de agosto de 2021.
Aceptado:
23 de setiembre de 2021.
*Correspondencia
María
Concepción Rocha-Arrieta Calle 75 A sur # 52f- 90, Cartagena, Colombia
Correo:
rochamariac201@gmail.com
Financiamiento
Trabajo
financiado por los autores.
Conflictos de interés
Los
autores declaran no tener ningún conflicto de interés.
Protección de sujetos
humanos y animales
Se
obtuvo el consentimiento informado por escrito del paciente para la publicación
de este informe de caso y todas las imágenes adjuntas.
Citar como:
Rocha-Arrieta
MC, Arias-Díaz A, Quiróz-Romero CA, Rocha-Arrieta Y.
Taquicardia ventricular polimórfica catecolaminérgica en adolescente: un
diagnóstico clínico, electrocardio- gráfico y
genético. Arch Peru Cardiol Cir Cardiovasc.
2021;2(3):205-210. doi: 10.47487/apcyccv.v2i3.151.