Artículo de Revisión
El adulto con tetralogía
de Fallot: lo que el cardiólogo clínico necesita saber
The adult
with tetralogy of fallot: what
the clinical cardiologist needs to know
Samantha Chávez-Saldívar1,2,a
https://orcid.org/0000-0002-8555-0305
Juan
Carlos Mego 1,2,a
https://orcid.org/0000-0002-3478-1832
Astrid
Tauma-Arrué1,2,a
https://orcid.org/0000-0003-2915-2158
Joel
Coronado1,2,a
https://orcid.org/0000-0001-9530-6683
Odalis
Luis-Ybáñez1,2,a
https://orcid.org/0000-0002-5009-4119
Katia
Bravo-Jaimes 3,b*
https://orcid.org/0000-0001-7745-7162
DOI:
https://doi.org/10.47487/apcyccv.v2i2.138
Resumen
La tetralogía de Fallot es la
cardiopatía congénita cianótica más frecuente. Luego de más de siete décadas desde
la primera cirugía paliativa, el pronóstico de esta enfermedad ha cambiado radicalmente.
Con la mayor sobrevida de pacientes con tetralogía de Fallot hacia la adultez,
el cardiólogo clínico enfrenta retos en el manejo de esta población, desde la regurgitación
pulmonar severa hasta la falla cardíaca y arritmias ventriculares. Su prevalencia
es aproximadamente 3 por cada 10 000 nacidos vivos, representando 7 a 10% de las
cardiopatías congénitas. En esta revisión se describirán los aspectos más
relevantes del cuidado de pacientes adultos con esta enfermedad.
Palabras clave: Tetralogía
de Fallot; Adultos; Sobrevida; Insuficiencia Cardíaca (fuente: DeCS BIREME).
Abstract
Tetralogy of Fallot (TOF) is the most common
cyanotic congenital heart disease. After more than seven decades
of the first
palliative surgery, TOF
prognosis has changed dramatically. The prevalence of TOF is approximately 3 per 10 000 births, representing 7 to 10% of congenital
heart disease. With a higher survival
into adulthood, the clinical cardiologist
faces challenges in the management of this
population, from severe pulmonary regurgitation to heart failure and ventricular arrhythmias. Its prevalence is approximately
3 per 10 000 live births, representing 7 to 10% of congenital heart
disease. This review will describe the most relevant
aspects of the care of
adult patients with this disease.
Keywords: Tetralogy of Fallot; Adults; Survival; Heart Failure (source: MeSH NLM).
n 1671, Dan Niels Stensen
describió el primer caso de un conjunto de lesiones anatómicas presentes en un
feto que incluían estenosis subpulmonar, comunicación
interventricular subaórtica y aorta cabalgante (1). Muchos otros anatomistas describieron casos
similares en las décadas posteriores, pero en 1888, Étienne-Louis Arthur Fallot
postuló que el origen embriológico común de todas estas lesiones se debía a la
desviación superoanterior del conus
o infundíbulo y bautizó a esta entidad como la «enfermedad azul», y que hoy en
día lleva su nombre (2). En 1944, Eileen Saxon, con
tan solo 14 meses de edad, se convirtió en la primera persona con tetralogía de
Fallot (TOF) en ser tratada quirúrgicamente mediante el uso de la primera
fístula sistémico-pulmonar ideada por Alfred Blalock,
Hellen Taussig y Vivien Thomas. En los siguientes
años se realizaron alrededor de cien operaciones anualmente, gracias a las
cuales se pudo comprobar que la sobrevivencia había aumentado hasta 64% en un
periodo de seguimiento de 15 años (2).
Han transcurrido más de tres siglos
desde sus primeras descripciones y en la actualidad la TOF continúa siendo una
de las cardiopatías congénitas cianóticas más importantes en la práctica clínica.
En esta revisión, se discutirán características clínicas, manejo quirúrgico y
los cuidados del adulto con TOF.
Características
generales
La TOF es la cardiopatía congénita cianótica
más común, caracterizada por la desviación anterior y superior
del conus
o infundíbulo subpulmonar en relación con la trabeculación septomarginal, causando
obstrucción del tracto de salida del ventrículo derecho (OTSVD), comunicación
interventricular subaórtica (generalmente única, de tipo
membranoso y no restrictiva, excepto en casos raros donde el margen ventricular
está protegido por el tejido accesorio de la válvula tricuspídea
o cuando la hipertrofia septal estrecha el defecto (3)), desviación del origen de
la aorta hacia la derecha e hipertrofia del ventrículo derecho. La prevalencia de
esta patología es aproximadamente de 3 por cada 10 000 nacimientos, lo cual representa
entre el 7 y 10% de todas las malformaciones cardíacas congénitas (3).
En países como España, la incidencia de
TOF es de aproximadamente 4,1 por cada 10 000 nacimientos (4). En Sudamérica se
encuentran valores diferentes, con una prevalencia en países como Brasil (5), Costa
Rica (6) y Colombia (7) de alrededor de 3,4; 2,3 y 0,4 por cada 10 000 nacidos
vivos, respectivamente. La variabilidad existente es parcialmente explicada por
la implementación del tamizaje neonatal para cardiopatías congénitas críticas por
medio de la oximetría de pulso (8).
En el caso de Perú, los datos
epidemiológicos varían según la región y su altitud, siendo menos frecuentes en
pacientes provenientes de una altitud mayor de 2260 m (9). En ausencia de un programa
de tamizaje neonatal ajustado a la altura a nivel nacional, es posible que la mayoría de los niños
detectados se presenten tardíamente y
tengan mayores posibilidades
de fallecer en ciudades de altura. En Iquitos se estima que la
incidencia de TOF es de 0,07 por cada mil nacidos vivos (10).Por
otro lado, en Lambayeque se encontró que la TOF constituye el 4,6% de las cardiopatías
congénitas en menores de 5 años (11).
En países desarrollados, se calcula que
alrededor de 97% de los pacientes con cardiopatías congénitas sobreviven a la adultez
(12). En el caso de la TOF, se estima que 79% de los pacientes que nacieron
entre la década de los 80 sobrevivieron a la adultez (13), en contraste con más
de 90% que nacieron en la primera década del 2000 (12). A pesar de no contar con
datos de sobrevida en pacientes con TOF en nuestro país, se ha reportado que hasta
el 60% de los pacientes con una cardiopatía congénita cianótica severa llega a sobrevivir
al primer año en centros de referencia (14).
Genética
La etiología de TOF es multifactorial y
alrededor del 25% de los pacientes presentan alguna alteración cromosómica.
Entre las más frecuente se encuentran la microdeleción de la región q11 del cromosoma
22 (15), seguido de la trisomía 21 (especialmente asociada con el canal atrioventricular) y menos frecuentemente las trisomías 18 y
13, así como el síndrome de CHARGE (16). Múltiples moléculas de señalización y factores
de transcripción, responsables en la cardiogénesis, han
sido implicados en la TOF, de los cuales los más descritos son TBX1, GATA4, NKS2.5,
JAG1, FOXC2, TBXa y TBX5 (17). Mutaciones en el gen JAG1,
causante del síndrome de Alagille, se relacionan a un
solapamiento clínico con los trastornos por deleción 22q11.2 y pueden causar una
TOF aislada (18). Asimismo, mutaciones del gen NKX2.5 son encontradas hasta en 4%
de los pacientes no sindrómicos con TOF.
El síndrome de deleción 22q11.2 no solo
está asociado con TOF, sino con otras cardiopatías congénitas que afectan al conotronco (interrupción del arco aórtico, tronco arterioso,
etc.) así como también características faciales, inmunodeficiencias e hipocalcemia
relacionadas con la hipoplasia o aplasia tímica
y paratiroidea (19). Es importante notar que en los pacientes con TOF que
tengan atresia pulmonar, válvula pulmonar ausente o estenosis valvular pulmonar,
la prevalencia de deleción 22q11.2 es mayor y, por ende, se recomienda hacer
estudios genéticos en estos casos. A pesar de que estos pacientes no tienen
menor sobrevida luego de la cirugía, tienen mayor riesgo de complicaciones posoperatorias
como falla cardíaca o respiratoria, intubación prolongada, estridor laríngeo y sepsis
(20).
El rol de la consejería genética en los pacientes con TOF sindrómicos es invaluable, ya que la identificación temprana del síndrome de deleción del 22q11.2 permitirá consejería a los padres, así como el manejo apropiado de los problemas del neurodesarrollo, clínicos, psiquiátricos y reproductivos que se puedan presentar a futuro. Adicionalmente, en pacientes con TOF no sindrómicos, es importante destacar su potencial hereditario, con un riesgo de recurrencia de aproximadamente 3% (21).existen casos con solo una intervención paliativa e, incluso, ningún tipo de intervención (25).
Presentación clínica
Durante el periodo neonatal
La presentación clínica de los pacientes
con TOF depende principalmente del grado de OTSVD, flujo pulmonar y sistémico (21).
Los neonatos cuya OTSVD sea leve o moderada, pueden lucir acianóticos,
tener un soplo sistólico detectable en el primer día de vida o presentar una leve
desaturación (85-90%). Por ello, esta presentación se denomina “TOF rosada” (22).
En casos de OTSVD leve y comunicación interventricular grande puede darse paso a
síntomas de insuficiencia cardiaca congestiva (23). Por otro lado, aquellos
neonatos con OTSVD severa suelen presentarse con hipoxemia significativa, con saturaciones
<80% como resultado del bajo flujo pulmonar y el gran shunt de derecha a izquierda.
Algunos infantes pueden presentar cuadros agudos
de cianosis llamados «episodios cianóticos», que son más frecuentes entre
los 2 a 4 meses de edad y son desencadenados generalmente por situaciones de estrés
como deshidratación, llanto, alimentación o defecación. En estos casos, el aumento
de la resistencia vascular pulmonar o la disminución de la resistencia vascular
periférica ocasiona un incremento del shunt de derecha a izquierda a través de
la comunicación interventricular, subsecuente hipoxemia, activación del centro respiratorio,
hiperventilación y aumento del tono adrenérgico, que puede ocasionar
empeoramiento de la OTSVD. Sin un tratamiento inmediato (llevar las rodillas
hacia el pecho o administrar fenilefrina para incrementar la resistencia
vascular periférica, dar un bolo de fluidos para aumentar la precarga del ventrículo
derecho, morfina y betabloqueantes endovenosos para aliviar la OTSVD), pueden conllevar
a arritmias, síncope y, ocasionalmente, a la muerte (1). Raramente, algunos infantes
pueden presentarse con abscesos cerebrales o convulsiones (19).
En 5% de los casos, la TOF puede
presentarse de manera extrema, con atresia pulmonar, ausencia completa de arterias
pulmonares nativas y colaterales aortopulmonares mayores (MAPCAs)
(24). Esta es una de las formas más severas de cardiopatía congénita, ya que tiene
un curso clínico que frecuentemente tiene como complicación el desarrollo de
hipertensión pulmonar.
Durante la adultez
La mayoría de los pacientes con TOF que
se presentan en la adultez han tenido una reparación completa, pero también progresiva
al ejercicio por lo que el seguimiento clínico es necesario en estos pacientes (26,27).
En pacientes con reparación completa
de TOF, la principal secuela hemodinámica es la insuficiencia valvular pulmonar
crónica, la cual condiciona una dilatación del ventrículo derecho y una
posterior disfunción sistólica del mismo, ocasionando insuficiencia cardiaca,
arritmias ventriculares e intolerancia
Sin intervención quirúrgica durante la
infancia o adolescencia, se pueden encontrar pacientes adultos con TOF en la adultez
temprana; sin embargo, menos del 5% sobreviven más allá de la cuarta década de
la vida (28). Aquellos que sobreviven, generalmente tienen un buen balance entre
el flujo a través de la comunicación interventricular y el grado de estenosis pulmonar.
Esta última protege la circulación pulmonar del alto flujo producto del shunt interventricular,
pero no es lo suficientemente obstructiva como para causar cianosis. Otros pacientes
pueden tener ya síndrome de Eisenmenger debido a una comunicación interventricular
no restrictiva y estenosis pulmonar mínima (25). Clínicamente estos pacientes
presentan cuadros de disnea y baja tolerancia al ejercicio. También como
consecuencia de un estado de cianosis crónica pueden desarrollar complicaciones
como policitemia, hiperviscosidad, acropaquias, alteraciones
de la hemostasia, abscesos cerebrales y endocarditis (22).
Manejo médico y
quirúrgico
Una vez establecido el diagnóstico
usando ecocardiografía Doppler, se tendrá una estimación del flujo pulmonar y la
necesidad de intervención temprana. En aquellos pacientes con presentaciones muy
severas (atresia pulmonar), la ausencia de comunicación entre el ventrículo derecho
y la arteria pulmonar, resulta necesaria la comunicación entre la circulación pulmonar
y sistémica. Para ello se hace uso de prostaglandina E, la cual permite la patencia
del ductus arterioso, mejorando así la presión arterial de oxígeno hasta que se
considere oportuno el abordaje quirúrgico. En casos menos severos, donde
predomina la estenosis subpulmonar, el empleo de bloqueantes
de los receptores beta-adrenérgicos previenen episodios de hipercianosis
antes de que se realice la cirugía (29), sin embargo, debido al potencial bloqueo
de la respuesta adrenérgica luego la cirugía su uso es heterogéneo (30).
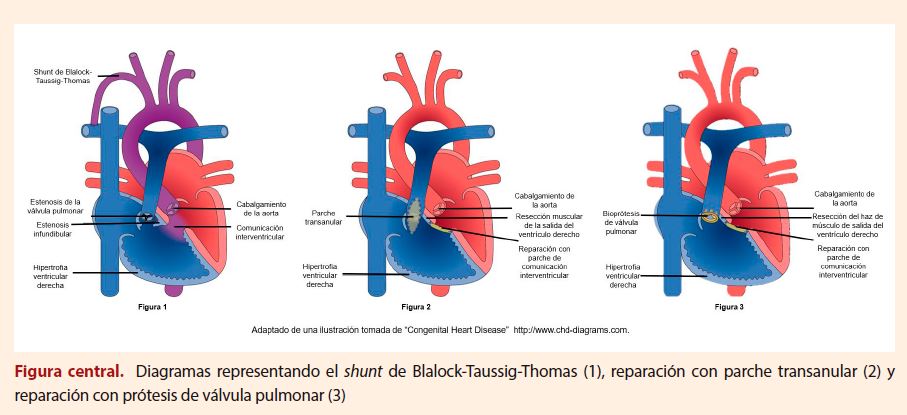
El manejo quirúrgico de los pacientes con
TOF ha evolucionado con el tiempo (Figura central). En aquellos pacientes que debutan con cianosis
muy severas desde el nacimiento pueden realizarse cirugías paliativas en un primer
momento como el shunt de Blalock-Taussig-Thomas
que consiste en anastomosar la arteria subclavia con la arteria pulmonar con la
finalidad de redistribuir el flujo sanguíneo hacia los pulmones, la cual se ha visto
mermada por la OTSVD. El procedimiento de Blalock-Taussig-Thomas modificado, consiste en la interposición de
un injerto entre la arteria subclavia y su arteria pulmonar ipsilateral (31). Gracias a los
avances en cateterismo cardíaco y procedimientos transcatéter
también puede realizarse la colocación de un stent en
el ductus arterioso, permitiendo un shunt sistémico-pulmonar (32).
Adicionalmente, la colocación de un stent en el TSVD ha demostrado tener un rol estabilizador en
infantes con múltiples comorbilidades que no son candidatos a cirugía temprana (33).
Debido a la diversidad de presentaciones
clínicas, el tiempo de realización de los procedimientos quirúrgicos en estos pacientes
también es variable. En general, la mayoría de infantes se somete a cirugía correctiva
entre los 3 a 6 meses de vida (34), ya que existe un mayor riesgo de estancia prolongada
hospitalaria así como en la unidad de cuidados
intensivos en aquellos pacientes menores de 3 meses (35); sin embargo, es posible
llevarla a cabo en centros altamente especializados.
La
cirugía reparadora involucra el cierre del defecto septal ventricular junto al manejo
de la OTSVD. Esta última puede realizarse con:
·
Valvulotomía pulmonar.
·
Resección del músculo infundibular.
·
Colocación de un parche transanular:
conlleva a insuficiencia valvular pulmonar severa debido a la disrupción del anillo
(36).
·
Colocación de un parche en el tracto de salida
del ventrículo derecho sin alterar la válvula pulmonar (valve-sparing).
·
Implantación protésica de una válvula
pulmonar (generalmente en adultos).
Cerca de 25% de pacientes muere en el primer
año de vida si no recibe cirugía. A mayor edad al momento de la cirugía reparadora,
la sobrevida tardía es menor. Así, la historia natural de la TOF demuestra que,
en ausencia de cirugía, 40% de pacientes muere antes de los 3 años de vida, 70%
antes de los 10 años y 95% antes de los 40 años (3). Sin embargo, si una persona
adulta es diagnosticada con TOF, la reparación quirúrgica tardía mejora los resultados
a largo plazo. Los pacientes que se presentan durante la adultez pueden tener
presiones arteriales pulmonares elevadas a pesar de tener OTSVD severa debido a
la cianosis crónica o a la disfunción ventricular izquierda. Sin embargo, esto no
debería excluirlos de una reparación quirúrgica debido a que el ventrículo derecho ha estado
sometido a presiones sistémicas desde el nacimiento (debido a la comunicación interventricular) y está preparado para la hipertensión pulmonar
posoperatoria.
La lesión pulmonar por
reperfusión que suele
suceder luego de
la reparación tardía
se presenta como
edema pulmonar en pacientes
adultos con cianosis marcada y flujo pulmonar restrictivo. Afortunadamente esta
suele ceder luego de días bajo ventilación a presión positiva.
Cuidados del adulto
con TOF
Regurgitación
pulmonar severa
Una de las complicaciones tardías más frecuentes
de los pacientes con reparación quirúrgica de TOF es la regurgitación pulmonar.
Por lo menos un tercio de los casos de TOF sometidos a cirugía reparadora durante
la infancia perduran con insuficiencia pulmonar severa (37). Esta lesión
hemodinámica es tolerada por muchos años; sin embargo, a la larga, genera
dilatación anular y ventricular derecha, así como disfunción sistólica del mismo, desencadenando
intolerancia al ejercicio, falla ventricular
derecha, arritmias y muerte súbita (38). Esto se refleja en un incremento del riesgo
de muerte en la tercera década posterior a la cirugía de 0,27 a 0,94% anual (39,40).
La ecocardiografía es una herramienta útil
para detectar la regurgitación pulmonar severa; sin embargo, debido a la forma tridimensional
del ventrículo derecho, tanto el volumen como la función de este son mejor evaluados
con resonancia magnética cardíaca. Este método no invasivo no depende de la ventana
acústica y se ha posicionado como el más exacto para determinar el tamaño y la función
ventricular derechos, los cuales son útiles en el seguimiento a largo plazo, por
lo cual se recomienda su obtención aproximadamente cada 3 años (41).
A través de la resonancia magnética
cardíaca, además de evaluar los volúmenes y funciones, también se observa la
viabilidad miocárdica, con lo cual es posible evidenciar fibrosis y vislumbrar la
posibilidad de futuras arritmias. Además, permite la delineación del curso de las
arterias coronarias, ya que 5 a 7% de pacientes presentan una coronaria
izquierda anómala que cruza el tracto de salida del ventrículo derecho que puede
ser comprimida en el momento de una intervención. Asimismo, permite evaluar anatómicamente
las ramas derecha e izquierda de la arteria pulmonar y delinear el flujo pulmonar
individual (41).
El reemplazo valvular pulmonar puede ser quirúrgico
o vía transcatéter. Ambos conllevan un
bajo riesgo operatorio en general, y puede llevar a la mejora
de síntomas, así como de remodelamiento reverso del ventrículo derecho
(disminución del volumen del ventrículo derecho, el cual se ha visto que
puede regresar a niveles basales luego de 7 a 10 años luego del reemplazo valvular
pulmonar). A pesar de estos resultados favorables, aún no hay evidencia de que el
reemplazo valvular pulmonar conlleve a una mejora de la función sistólica ventricular
derecha, prevención de arritmias o disminuya la mortalidad (41). Asimismo, el tiempo
óptimo para el reemplazo valvular pulmonar es sujeto de controversia, debido a que
se necesita poner en una balanza por un lado los riesgos asociados con la regurgitación
pulmonar severa y, por el otro, la finita duración de las válvulas bioprotésicas (tomando en cuenta que las válvulas mecánicas
en la posición pulmonar se asocian con un altísimo riesgo de trombosis y falla temprana,
su uso no es recomendado actualmente). Las indicaciones actuales, de acuerdo con
las guías del American College of
Cardiology/American Heart Association
del 2018 (42) se encuentran en la Tabla 1. A pesar del énfasis en los volúmenes ventriculares
derechos, se debe resaltar que estos no siempre predicen su normalización luego
del reemplazo valvular pulmonar. Asimismo, la incapacidad del tamaño ventricular
derecho luego del reemplazo valvular pulmonar para predecir resultados clínicos
adversos en estos pacientes resalta la necesidad de desarrollar nuevos criterios
a futuro.
En el año 2000 se utilizó por primera
vez una válvula venosa yugular bovina montada en un stent
(válvula Melody) para el tratamiento de la regurgitación pulmonar severa y estenosis
residual luego de la implantación de una válvula bioprotésica
quirúrgica. Con el paso de los años también se ha hecho uso de esta válvula en TSVD
nativos que cuenten con un diámetro adecuado. El mayor diámetro de las válvulas
Sapien S3 (que se utilizan en los reemplazos de válvulas
aórticas vía transcateter [TAVR]) hace posible su uso
en ausencia de bioprótesis donde el anillo pulmonar no se encuentre severamente
dilatado, y efectuado en centros altamente especializados (43). Es importante
resaltar el rol de la profilaxis antibiótica dental en pacientes con bioprótesis
u homoinjerto pulmonar, ya que el riesgo de
endocarditis infecciosa es significativo.
Comunicación
interventricular residual
La mayoría de reoperaciones
de estos defectos están relacionadas con las fugas residuales a través del parche
de comunicación interventricular. Debido al riesgo de disfunción del ventrículo izquierdo,
su reintervención está indicada por medio de cierre con sutura o parche si el Qp/Qs es mayor o igual de 1,5:1, si
la presión sistólica pulmonar no excede los dos tercios de la presión sistólica
sistémica y la resistencia vascular pulmonar es menor a dos tercios de la resistencia
vascular sistémica. Asimismo, está indicada la reintervención si el paciente requiere
cirugía cardíaca por otros motivos. Si la comunicación interventricular residual
no se encuentra cerca del nodo atrioventricular o de las
válvulas tricúspide o aórtica, es posible cerrar el defecto vía transcatéter bajo guía de ecocardiografía transesofágica. Es
importante el uso de antibioticoterapia profiláctica para prevenir endocarditis
infecciosa en pacientes que presenten estos defectos residuales luego de la cirugía
reparadora así como también en aquellos con cirugías paliativas
y los no reparados (3).

Dilatación de la
aorta
La dilatación de la raíz de la aorta o
de la aorta ascendente está presente entre el 15-88 % de pacientes con TOF (44)
y en el 20% de pacientes con TOF reparada (45). Los factores de riesgo
relacionados con una mayor incidencia son el sexo masculino, patología aórtica congénita
y la cirugía reparadora en la edad adulta (45). Las manifestaciones clínicas son
diversas en cuanto a
variabilidad y gravedad, siendo las principales; regurgitación valvular aórtica,
aneurisma de aorta ascendente y disección
de aorta. De esta última, se asume que es una complicación sumamente rara, ya que
de 18 353 hospitalizaciones en pacientes con TOF (provenientes del National Inpatient Sample de Estados Unidos), solo 11 estuvieron relacionadas con
disección aórtica (0,06%). A pesar de su rareza, la identificación oportuna es
esencial ya que la mortalidad alcanzó al 45% de los pacientes mencionados en dicho
estudio (46).
Falla cardíaca
La insuficiencia cardiaca en pacientes
con TOF se presenta a una edad más joven en comparación a la población general,
con una edad promedio de aparición de 50 años y siendo más frecuente en varones
(30). La disfunción ventricular no es exclusiva del ventrículo derecho, ya que también
puede ser biventricular debido a interacciones
interventriculares. Así, la dilatación de la pared del ventrículo derecho
desplaza el septum interventricular hacia el ventrículo izquierdo, alterando su
rotación apical y, finalmente, produciendo desincronización interventricular y disfunción
sistólica del ventrículo izquierdo (47,48). Otros mecanismos fisiopatológicos como
las concentraciones bajas de oxígeno, sobrecarga de presión (48), lesión hipóxica
isquémica y la regurgitación valvular pulmonar residual posquirúrgica, pueden
ocasionar estiramiento de las fibras miocárdicas así como
circuitos de reentrada alrededor de las cicatrices de ventriculotomía
(49-51), los cuales degeneran en limitación del gasto cardiaco y taquiarritmias.
El manejo en pacientes adultos con TOF
y falla cardíaca es extrapolado de la población en general (52). La poca evidencia
específica en esta población en el ensayo REDEFINE no logró demostrar ningún
efecto beneficioso del losartán sobre la función
ventricular derecha (53). Si el tratamiento médico no es suficiente y, por lo contrario,
la falla cardíaca progresa (54), es necesario considerar el trasplante cardiaco,
resaltando que en general, la sobrevida de los pacientes con cardiopatías congénitas
luego del primer año postransplante es mayor a la de aquellos
transplantados por causas no relacionadas a cardiopatías congénitas
(55).
Arritmias
ventriculares
Aproximadamente 10% de los adultos con
TOF reparada desarrollan taquicardia ventricular sostenida, y el riesgo aumenta
conforme la edad avanza. La identificación de los pacientes que se encuentren en
mayor riesgo de presentar arritmias ventriculares se hace por medio del Holter
(mayor sensibilidad con el uso de uno a largo plazo), el cual está indicado en aquellos
pacientes sintomáticos o con lesiones residuales significativas en un periodo anual
o bianual. Los factores de riesgo de arritmias ventriculares y muerte súbita incluye la duración del complejo QRS mayor a 180 ms (y sobre
todo la fragmentación del complejo QRS), disfunción sistólica o diastólica
ventricular derecha o izquierda, historia de taquicardia ventricular, hipertrofia
ventricular derecha o cicatriz extensa vista en resonancia magnética cardíaca, taquicardia
ventricular inducible o sustrato de alto riesgo visto en un estudio
electrofisiológico (41,56-58). De otro lado, datos provenientes del riesgo de shocks
apropiados en pacientes con TOF y desfibrilador automático implantable, nos permite
identificar aquellos que puedan beneficiarse de este dispositivo como prevención
primaria (Tabla 2) (59). La decisión de implantar un desfibrilador automático
implantable siempre debe ser individualizada, luego de tener una discusión acerca
de riesgos y beneficios con el paciente, ya que a pesar de la tasa anualizada de
shocks apropiados es alta, también existe riesgo a largo plazo de complicaciones
de los electrodos, así como shocks inapropiados.
Manejo durante el
embarazo
El embarazo constituye un periodo de alto
riesgo fetal para pacientes con TOF no reparada, principalmente por la cianosis
(sobre todo en aquellas pacientes con saturación de oxígeno menor a 85%). La baja
resistencia vascular periférica que se da para dar mayor flujo uterino ocasiona
un aumento del shunt de derecha a izquierda, empeorando la cianosis y generando
una tasa de hasta 30% de óbitos fetales y 4% de muerte materna.
En pacientes con TOF reparada quirúrgicamente,
el riesgo asociado con el embarazo depende del estado hemodinámico actual de la
paciente. Si existen lesiones residuales, OTSVD, regurgitación pulmonar severa o
falla ventricular derecha, e incremento de la precarga de 50% propio del embarazo
pueden generar falla cardíaca o arritmias.

Se recomienda que todas las pacientes con
TOF tengan consejería cardiológica prenatal, ecocardiografía fetal a las 20
semanas de gestación, monitoreo cardiológico y ecocardiográfico frecuente (al menos
en cada trimestre de la gestación y en el período posparto temprano), así como un
plan de parto.
Seguimiento continuo
Debido a los avances en el diagnóstico
y tratamiento de la TOF se reporta una supervivencia del 90% de los individuos en
países desarrollados. Sin embargo, esta prolongación de años no necesariamente
significa una ganancia de calidad de vida. Para lograr una calidad de vida adecuada
se recomienda enfatizar no solo el cuidado físico sino el socioemocional y neurobiológico.
Es importante el seguimiento médico no solo en caso de síntomas agudos, sino para
tener una información continua de la buena salud del paciente (60) e identificar
áreas donde se requiera mayor educación, recursos sociales o apoyo de otros profesionales.
Un aspecto esencial del cuidado de adultos con cardiopatías
congénitas es la planificación anticipada de los cuidados, la cual brinda
beneficios a la calidad de vida y una atención médica concordante con los objetivos
del paciente (61). Muchos factores como el costo de crear documentos legales, la
falta de recursos para tomar decisiones informadas, la negación y evitación
hacen que estos objetivos del cuidado no sean discutidos durante la visita
médica. Sin embargo, tomarse un tiempo para facilitar esta conversación podría aliviar
la carga de los seres queridos y garantizar que se respeten las preferencias del
paciente (62).
Conclusiones
Los pacientes con tetralogía de Fallot
requieren un cuidado longitudinal, inclusive luego de la reparación quirúrgica.
La regurgitación pulmonar severa, falla
cardíaca y arritmias ventriculares son complicaciones frecuentes en adultos con
tetralogía de Fallot reparada quirúrgicamente.
El objetivo del equipo médico a cargo de
estos pacientes no solo debe estar ligado a aumentar la esperanza de vida sino la
calidad de la misma.
Agradecimientos
A
la Dra. Diana Torpoco Rivera, por su revisión crítica del manuscrito.
Referencias
bibliográficas
1. Diaz-Frias J, Guillaume M. Tetralogy of Fallot. In: StatPearls. Treasure Island (FL): StatPearls
Publishing; 2020. p. 22.
2. Taussig HB. Neuhauser Lecture: Tetralogy of Fallot: early history and late results. AJR Am J Rroentgenol. 1979;133(3):422-31.
doi: 10.2214/ ajr.133.3.422.
3. Babu-Narayan S, Gatzoulis MA. Tetralogy of Fallot. In: Diagnosis and Management of
Adult Congenital Heart Disease. 3rd Edition. Elsevier;
2018.
4. Pérez- Picarzo J, Mosquera M, Latasa P, Crespo D. Incidence and evolution of congenital heart
disease in Spain from 2003 until 2012. An Pediatr (Engl Ed).
2018;89(5):294-301. doi: 10.1016/j.anpedi.2017.12.009.
5. Pinto V, Branco
K, Cavalcante R, Waldemiro J,
Costa J, Maria de Freitas S, et al. Epidemiology of congenital heart
disease in Brazil. Rev Bras Cir
Cardiovasc. 2015;30:219-24. doi: 10.5935/1678-9741.20150018.
6. Benavides A, Faerron JE, Umana L, Romero JJ. Epidemiología
y registro de las cardiopatías congénitas en Costa Rica. Rev
Panam Salud Publica. 2011;30:31-8.
doi:10.1590/S1020-49892011000700005.
7. Tassinari S, Martínez-Vernaza
S, Erazo-Morera N, Pinzón-Arciniegas MC, Gracia G, Zarante
I. Epidemiología de las cardiopatías congénitas en Bogotá, Colombia en el período
comprendido entre 2001 y 2014:
8. ¿Mejoría en la
vigilancia o aumento en la prevalencia? Biomedica.
2018;38(Sup1):141-8. doi: 10.7705/biomedica.v38i0.3381.
9. Martin GR, Ewer AK, Gaviglio A, Hom L , Saarinen A, Sontag M, et al. Updated Strategies for Pulse Oximetry Screening for Critical Congenital Heart Disease. Pediatrics. 2020;146(1):e20191650. doi:
10.1542/peds.2019-1650.
10. Ayasta A. Hinostroza
C. Asociación entre altura y cardiopatías congénitas en pacientes pediátricos en
el Instituto Nacional de Salud del Niño (INSN), Lima-Perú, en los años 2017-2018.
Lima; INSN; 2018.
11. Uribe J, Ramal
C, Olórtegui A, Pisconte C,
Elgegren J, Fritas R, et al. Incidencia de
cardiopatías congénitas en Iquitos, Perú. Rev Peru Cardiol. 2010;36(1):14-20.
12. Uribe AK, Díaz-Vélez
C, Cerrón-Rivera C. Características epidemiológicas y clínicas de las cardiopatías
congénitas en menores de 5 años del Hospital Almanzor Aguinaga Asenjo: Enero - Diciembre 2012. Horiz Med (Barcelona). 2015;15(1):49-56.
13. Mandalenakis Z, Giang KW, Eriksson P, Liden H, Synnergren M, Wåhlander H, et al. Survival in Children With Congenital
Heart Disease: Have We Reached a Peak
at 97%? J Am Heart Assoc. 2020;9(22):e017704.
doi: 10.1161/JAHA.120.017704.
14. Moons P, Bovijn L, Budts W, Belmans A, Gewillig M. Temporal trends in survival to adulthood among
patients born with congenital heart disease from
1970 to 1992 in Belgium. Circulation. 2010;122(22):2264- 72. doi:
10.1161/CIRCULATIONAHA.110.946343.
15. Torres-Romucho CE, Uriondo-Ore VG, Ramirez-Palomino AJ, Arroyo-
Hernández H, Valverde M , Protzel-Pinedo
A, et al. Factors associated
with survival at one year of
life in neonates with severe congenital
cardiopathy in A National Hospital
in Peru. Rev Perú Med Exp Salud Publica.
2019;36(3):433-441. doi: 10.17843/rpmesp.2019.363.4166.
16. Villafane J, Feinstein JA,
Jenkins KJ, Vincent R, Walsh E, Dubin A, et al. Hot topics in tetralogy of Fallot. J Am Coll Cardiol. 2013;62(23):2155-
66. doi: 10.1016/j.jacc.2013.07.100.
17. Oliveira PHA, Souza
BS, Pacheco EN, Menegazzo M, Corrêa
I, Zen P, et al. Genetic Syndromes
Associated with Congenital Cardiac Defects and Ophthalmologic Changes - Systematization for Diagnosis in the Clinical Practice. Arq Bras Cardiol.
2018;110(1):84-90. doi: 10.5935/ abc.20180013.
18. Morgenthau A, Frishman WH. Genetic Origins of Tetralogy
of Fallot. Cardiol Rev. 2018;26(2):86-92.
doi: 10.1097/CRD.0000000000000170.
19. Glaeser C, Kotzot D, Caliebe A, Kottke R, Schulz S, Schweigman U,
et al. Gene symbol: JAG1. Disease: tetralogy of Fallot. Hum Gen. 2006;119(6):674.
20. Rauch R, Hofbeck M, Zweier C , Koch A, Zink S, Trautmann U, et
al. Comprehensive genotype-phenotype
analysis in 230 patients with tetralogy of Fallot. J Med Genet.
2010;47(5):321-31. doi: 10.1136/ jmg.2009.070391.
21. Mahle WT, Crisalli J, Coleman K, Campbell R, Tam
V, Vincent R, et al. Deletion of
chromosome 22q11.2 and outcome
in patients with pulmonary atresia and ventricular septal defect. Ann Thorac Surg. 2003;76(2):567-71. doi: 10.1016/s0003-4975(03)00516-2.
22. Bailliard F, Anderson
RH. Tetralogy of Fallot. Orphanet J Rare Dis. 2009;4:2. doi:
10.1186/1750-1172-4-2.
23. Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts.
N Engl J Med. 2000;342(5):334-42. doi:
10.1056/ NEJM200002033420507.
24. Wise-Faberowski L, Asija R, McElhinney DB. Tetralogy of Fallot: Everything you wanted to know
but were afraid to ask.
Paediatr Anaesth.
2019;29(5):475-482. doi: 10.1111/pan.13569.
25. Leonard H,
Derrick G, O’Sullivan J, Wren
C. Natural and unnatural history
of pulmonary atresia. Heart.
2000;84(5):499-503. doi: 10.1136/ heart.84.5.499.
26. Bhatt AB, Foster E,
Kuehl K, Alpert J, Brabeck S, Crumb S, et al. Congenital heart disease in the older adult: a scientific statement from the American Heart Association. Circulation. 2015;131(21):1884-
931. doi: 10.1161/CIR.0000000000000204.
27. Babu-Narayan SV, Diller GP,
Gheta RR , Bastin A, Karonis T, Li W, et al.
Clinical outcomes of surgical pulmonary
valve replacement after repair of tetralogy
of Fallot and potential prognostic value of preoperative cardiopulmonary exercise testing. Circulation. 2014;129(1):18-27. doi:
10.1161/CIRCULATIONAHA.113.001485.
28. Huehnergarth KV, Gurvitz M, Stout KK, Otto CM. Repaired
tetralogy of Fallot in the adult: monitoring
and management. Heart. 2008;94(12):1663-9. doi: 10.1136/hrt.2008.147249.
29. Shinebourne EA, Babu-Narayan SV, Carvalho JS. Tetralogy
of Fallot: from fetus to adult.
Heart. 2006;92(9):1353-9. doi: 10.1136/
hrt.2005.061143.
30. Garson A, Jr., Gillette
PC, McNamara DG. Propranolol: the
preferred palliation for tetralogy of
Fallot. Am J Cardiol. 1981;47(5):1098-104. doi: 10.1016/0002-9149(81)90219-8.
31. Barazzone C, Jaccard C,
Berner M, Dayer P, Rouge J, Oberhansli I, et
al. Propranolol treatment
in children with tetralogy of Fallot alters the response to isoprenaline after surgical repair. Br Heart J.
1988;60(2):156-61. doi: 10.1136/hrt.60.2.156.
32. De Leval MR, McKay R, Jones M, Stark J, Macartney
FJ. Modified Blalock-Taussig
shunt. Use of subclavian artery orifice as flow regulator in prosthetic systemic-pulmonary artery shunts. J Thorac Cardiovasc Surg. 1981;81(1):112-9.
33. Rehman R, Marhisham MC, Alwi M. Stenting the complex
patent ductus arteriosus in
tetralogy of Fallot with pulmonary atresia: challenges and outcomes. Future Cardiol. 2018;14(1):55-73. doi:
10.2217/fca-2017-0053.
34. Sandoval JP, Chaturvedi RR, Benson L, Morgan G , Van Arsdell G , Honjo O, et al. Right Ventricular
Outflow Tract Stenting in Tetralogy of Fallot Infants With Risk Factors
for Early Primary Repair. Circ Cardiovasc Interv. 2016;9(12):e003979. doi: 10.1161/ CIRCINTERVENTIONS.116.003979.
35. Apitz C, Webb GD, Redington AN. Tetralogy of Fallot. Lancet. 2009;374(9699):1462-71.
doi: 10.1016/S0140-6736(09)60657-7.
36. Van
Arsdell GS, Maharaj GS, Tom
J, Rao VK, Coles JG, Freedom RM, et al. What is the
optimal age for repair of
tetralogy of Fallot? Circulation. 2000;102(19 Suppl 3):III123-9. doi: 10.1161/01.cir.102.suppl_3.iii-123.
37. Mainwaring RD, Hanley FL. Tetralogy of Fallot Repair: How I Teach It. Ann Thorac Surg.
2016;102(6):1776-1781. doi: 10.1016/j.
athoracsur.2016.09.111.
38. Geva T, Sandweiss BM, Gauvreau K, Lock
JE, Powell AJ. Factors associated
with impaired clinical status in long-term survivors of tetralogy
of Fallot repair evaluated by magnetic
resonance imaging. J Am
Coll Cardiol. 2004;43(6):1068-74. doi:
10.1016/j.jacc.2003.10.045.
39. Bouzas B, Kilner PJ, Gatzoulis MA. Pulmonary regurgitation: not a benign lesion.
Eur Heart J. 2005;26(5):433-9. doi:
10.1093/eurheartj/ ehi091.